Huntington
A culpa deste «post» altamente didáctico e pouco jocoso é fruto duma conversa que eu tive no LAF! com o Miguel Barros após a sua actuação.
Foi ele que me alertou acerca desta doença (que apesar de não ser tão 'popular' como Parkinson ou Alzheimer, também é grave) e da relevância que a utilização de células estaminais podem ter no seu combate.
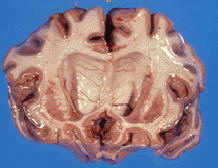
Huntington é uma doença hereditária, degenerativa, do foro neurológico que leva a demência. Todos os sintomas da doença Huntington são devidos a uma pequena mudança no gene da huntingtin no cromossoma 4.
Quem tem a doença tem um gene huntingtin (HD) ligeiramente maior que o usual. Tal acontece porque três letras do código genético CAG são repetidas muitas vezes. O número de repetições CAG varia de pessoa para pessoa. Um indíviduo saudável tem entre 9 - 35 repetições. Pessoas com a doença têm entre 36 - 121 repetições. Um excesso de repetições CAG muda a forma da proteína chamada huntingtina, produzida pelo gene.
Cada repetição CAG no gene existe para se fabricar um determinado aminoácido: a glutamina com a expansão poliglutaminica, a huntingtina mutada fica com outra configuração.
Com esta nova configuração a huntingtina tem interacção com varias proteínas que seriam ignoradas caso a proteína fosse a normal; isto acontece sobretudo nos neurónios no cérebro. Dentro do neurónio uma proteína interactiva corta a huntingtina em duas partes e uma delas entra no nucleo. À medida que mais huntingtina mutada é clivada gera-se uma acumulação de residuos no núcleo que se começam a agregar. O papel destes agregados e se têm alguma influência na patologia ainda não está completamente descrito mas o que se observa é que os neurónios deixam de ter a sua total funcionalidade e acabam por morrer dando origem a todos os sintomas da doença.
O primeiro a classificar e descrever esta trágica doença foi Johan Christian Lund, um físico Norueguês que lhe deu o nome de "Chorea St Vitus" em 1860, mas esta descrição atraiu muito pouco a atenção da população e foi esquecida.
O que chamou verdadeiramente à atenção, foi a descrição dada pelo Dr. George Huntington em 1872 enquanto médico em Long Island, New York, que, apesar de não ser a primeira descrição, foi a mais precisa até então e especialmente interessante pela capacidade de preceder as teorias genéticas de Gregor Mendel que, só seriam redescobertas 30 anos depois.
George Huntington descreveu nas suas memórias uma rara doença que já o seu pai e avô (ambos médicos) haviam reconhecido em alguns dos seus pacientes. Assim, com base nas notas do seu pai e avô e também nas suas próprias observações, Huntington decidiu estudar e escrever sobre este fenómeno. Em sua homenagem, esta doença passou a ser conhecida como Coreia de Huntington. Actualmente, esta tomou o nome de doença de Huntington, porque a "coreia (origem da língua grega = dançar, movimentos involuntários) apenas descreve um dos sintomas da doença".
A HD "afecta as capacidades motoras individuais e também as capacidades intelectuais e emocionais". Sendo uma degeneração geneticamente programada das células do cérebro, a HD manifesta-se nas células do gânglio basal (estrutura importante na coordenação dos movimentos), especialmente os neurónios estriados de núcleo caudado. Outra parte do cérebro que também é afectada é o córtex, que controla o pensamento, a percepção e a memória.
O que chamou verdadeiramente à atenção, foi a descrição dada pelo Dr. George Huntington em 1872 enquanto médico em Long Island, New York, que, apesar de não ser a primeira descrição, foi a mais precisa até então e especialmente interessante pela capacidade de preceder as teorias genéticas de Gregor Mendel que, só seriam redescobertas 30 anos depois.
George Huntington descreveu nas suas memórias uma rara doença que já o seu pai e avô (ambos médicos) haviam reconhecido em alguns dos seus pacientes. Assim, com base nas notas do seu pai e avô e também nas suas próprias observações, Huntington decidiu estudar e escrever sobre este fenómeno. Em sua homenagem, esta doença passou a ser conhecida como Coreia de Huntington. Actualmente, esta tomou o nome de doença de Huntington, porque a "coreia (origem da língua grega = dançar, movimentos involuntários) apenas descreve um dos sintomas da doença".
A HD "afecta as capacidades motoras individuais e também as capacidades intelectuais e emocionais". Sendo uma degeneração geneticamente programada das células do cérebro, a HD manifesta-se nas células do gânglio basal (estrutura importante na coordenação dos movimentos), especialmente os neurónios estriados de núcleo caudado. Outra parte do cérebro que também é afectada é o córtex, que controla o pensamento, a percepção e a memória.
A HD encontra-se em qualquer país do mundo e o seu modo de transmissão é de pais para filhos com um risco de 50%, isto porque, se um progenitor é seu portador, qualquer criança seu descendente (sexo masculino ou feminino) tem 50% de possibilidades de herdar a doença.
Por isso se diz que esta doença é autossómica dominante, uma vez, que basta uma só cópia do gene defeituoso, herdado de um dos progenitores, para a doença se manifestar.
"Os primeiros sintomas podem surgir no indivíduo por volta dos 30-45 anos; apenas em 5% das pessoas aparecem sintomas antes dos 20 anos (normalmente conhecida como HD juvenil) e 5% não mostram sintomas até terem 60 anos. Depois dos 70 anos o risco de aparecimento da doença é muito reduzido."
A origem desta doença encontra-se numa alteração de um gene, cuja localização foi descrita em 1983. A partir dos trabalhos de muitas equipas de cientistas liderados por James Gusella, foi estabelecido que o gene da HD se encontra em linkage com a zona RFLP localizada no braço pequeno do cromossoma 4.
Em 1993, através de uma procura internacional, descobriu-se a localização exacta deste gene (cromossoma 4p16.3 - que se manteve desconhecida durante 10 anos devido à sua próxima localização da extremidade do braço pequeno do cromossoma 4. Assim, depois de se clonar e sequenciar o gene mutante, detectou-se que esta mutação era uma expansão característica do tripleto CAG (citosina, adenina e guanina) que dá origem ao aminoácido glutamina. Portanto, este gene que inicialmente codificava a proteína "huntingtina", quando mutado, dá origem a uma outra proteína (com uma maior extensão de poliglutamina que o normal) que, apesar de levar a cabo as suas funções normais de forma correcta, também realiza outras funções que causam esta doença. Desconhecem-se algumas das funções da proteína huntingtina "normal", mas parece que estão relacionadas com os orgãos e com as funções de transporte. Na HD, a proteína "pega-se" até formar glóbulos que são encontrados nos cérebros de pacientes com esta patologia. Esta proteína defeituosa vai-se acumulando nos neurónios de certas regiões do cérebro, fazendo com que estes deixem de trabalhar devido ao seu efeito tóxico. "As doenças neurodegenerativas acabam por matar um grande número de neurónios, mas os seus gravíssimos sintomas começam a manifestar-se muito antes, durante um período muito prolongado em que os neurónios continuam vivos mas não funcionam." "Estudos deste gene em 75 famílias de HD revelam que os indivíduos não afectados possuem 11 a 38 cópias deste tripleto (CAG), enquanto que os membros afectados têm desde 42 a mais de 66 cópias. Quantas mais cópias presentes mais severos são os sintomas e mais cedo estes aparecem, com as mais longas repetições do tripleto encontradas em juvenis com HD. (O caso mais extremo foi o de um alelo com, aproximadamente 100 cópias, de uma criança em que a HD se começou a manifestar quando esta tinha apenas 2 anos). Normalmente, o número de tripletos aumenta de geração em geração, mas ocasionalmente, existem diminuições deste numero." "Parece ter-se notado que as pessoas com HD juvenil normalmente, haviam herdado esta doença do seu pai. Tendo também tendência para ter um número maior de repetições de CAG. Os cientistas crêem que este facto se deve ao processo de produção do esperma. Ao contrário dos óvulos, o esperma é produzido aos milhões. Dado que o DNA é copiado milhões de vezes durante o processo, pensa-se que existem mais possibilidades para que ocorra um erro genético".
Em 1993, através de uma procura internacional, descobriu-se a localização exacta deste gene (cromossoma 4p16.3 - que se manteve desconhecida durante 10 anos devido à sua próxima localização da extremidade do braço pequeno do cromossoma 4. Assim, depois de se clonar e sequenciar o gene mutante, detectou-se que esta mutação era uma expansão característica do tripleto CAG (citosina, adenina e guanina) que dá origem ao aminoácido glutamina. Portanto, este gene que inicialmente codificava a proteína "huntingtina", quando mutado, dá origem a uma outra proteína (com uma maior extensão de poliglutamina que o normal) que, apesar de levar a cabo as suas funções normais de forma correcta, também realiza outras funções que causam esta doença. Desconhecem-se algumas das funções da proteína huntingtina "normal", mas parece que estão relacionadas com os orgãos e com as funções de transporte. Na HD, a proteína "pega-se" até formar glóbulos que são encontrados nos cérebros de pacientes com esta patologia. Esta proteína defeituosa vai-se acumulando nos neurónios de certas regiões do cérebro, fazendo com que estes deixem de trabalhar devido ao seu efeito tóxico. "As doenças neurodegenerativas acabam por matar um grande número de neurónios, mas os seus gravíssimos sintomas começam a manifestar-se muito antes, durante um período muito prolongado em que os neurónios continuam vivos mas não funcionam." "Estudos deste gene em 75 famílias de HD revelam que os indivíduos não afectados possuem 11 a 38 cópias deste tripleto (CAG), enquanto que os membros afectados têm desde 42 a mais de 66 cópias. Quantas mais cópias presentes mais severos são os sintomas e mais cedo estes aparecem, com as mais longas repetições do tripleto encontradas em juvenis com HD. (O caso mais extremo foi o de um alelo com, aproximadamente 100 cópias, de uma criança em que a HD se começou a manifestar quando esta tinha apenas 2 anos). Normalmente, o número de tripletos aumenta de geração em geração, mas ocasionalmente, existem diminuições deste numero." "Parece ter-se notado que as pessoas com HD juvenil normalmente, haviam herdado esta doença do seu pai. Tendo também tendência para ter um número maior de repetições de CAG. Os cientistas crêem que este facto se deve ao processo de produção do esperma. Ao contrário dos óvulos, o esperma é produzido aos milhões. Dado que o DNA é copiado milhões de vezes durante o processo, pensa-se que existem mais possibilidades para que ocorra um erro genético".
Os primeiros sintomas desta doença variam muito de pessoa para pessoa mas, o que é comum entre todos os seus pacientes é que quanto mais cedo os seus sintomas aparecem mais rapidamente a doença progride. A HD causa movimentos incontrolados e descoordenados, perda de equilíbrio, memória e de concentração, depressão, mudança de humor, irritação e apatia. Estes sintomas podem desaparecer com a avanço da doença ou podem continuar e incluir mais explosões de hostilidade e profundos ataques de depressão. "A HD pode afectar o raciocínio, a memória, e outras funções cognitivas. Os primeiros sintomas podem incluir dificuldade em conduzir, em aprender coisas novas, responder a perguntas, tomar decisões." Os seus portadores podem ter graves problemas a andar, caindo muitas vezes. Com a progressão da HD, a linguagem pode tornar-se incompreensível, as funções vitais como comer, falar podem piorar bastante e, algumas pessoas podem chegar a não conhecer outras.
A HD (perturbação neurológica progressiva) cujo aparecimento se verifica normalmente em idade adulta, não possui nenhum tratamento efectivo e a morte chega 10 a 15 anos depois do aparecimento dos sintomas. Cerca de 60% dos casos são diagnosticados entre os 35 e os 50 anos. "Assim, pessoas com o alelo defeituoso podem reproduzir-se antes da ocorrência dos sintomas e, então os seus filhos têm de viver com a incerteza de serem ou não portadores desse alelo e, portanto se possuirão ou não a HD."
"Actualmente, a análise do DNA pode revelar a presença ou ausência do gene causador desta doença antes do começo dos sintomas. Em pessoas em risco, a decisão de realizar ou não o teste, pode ser um problema muito agonizante e angustiante."
"A selecção natural contra o gene da doença de Huntington não é forte até ao seu aparecimento tardio, o que significa, que a fitness biológica (reprodução diferencial) não afecta grandemente os heterozigóticos (portadores da doença). De facto, alguns estudos actuais, revelaram que os pacientes da doença de Huntington têm, em média, famílias numerosas. Se isto é geralmente verdade, com o passar de muitas gerações, a consequência teórica é que o gene da HD irá substituir o alelo normal e tornar-se -á o novo alelo normal nesse locus."
"A selecção natural contra o gene da doença de Huntington não é forte até ao seu aparecimento tardio, o que significa, que a fitness biológica (reprodução diferencial) não afecta grandemente os heterozigóticos (portadores da doença). De facto, alguns estudos actuais, revelaram que os pacientes da doença de Huntington têm, em média, famílias numerosas. Se isto é geralmente verdade, com o passar de muitas gerações, a consequência teórica é que o gene da HD irá substituir o alelo normal e tornar-se -á o novo alelo normal nesse locus."
Pronto, Miguel se leres isto, aqui fica o meu contributo para a divulgação de informação sobre a Doença de Huntington.
Um abraço para ti e um beijinho para a Susana. Feliz Natal.
publicado por Inês Ramos @ 4:20:00 a.m.
![]()


7 Comments:
Brrrr! Parkinson?? Tremo só de pensar nisso! Alzheimer?? Prefiro nem me lembrar disso!! Huntington?? Fico doido com medo!
Já agora...e em jeito de crítica...não me parece bem que alguém, estando em Santarém, não dê um salto a Leiria!!
Goth, se der tempo vamos tomar um café!
Fico á espera. Se não houver tempo para Goth Mortens não haverá tempo para o resto do mundo.
Ahfuedasse!
Agora fiquei toda babada...
«...Os investigadores acreditam que as células estaminais oferecem enormes potencialidades para a reparação de tecidos doentes ou danificados, substituindo as células "fracas" por novas. "As células estaminais oferecem promessas reais para o tratamento de doenças actualmente incuráveis", declarou o professor Colin Blakemore, chefe-executivo do Conselho de Investigação Médica...»
Ler o artigo todo em:
http://ultimahora.publico.pt/shownews.asp?id=1194272
Este Natal, haja esperança... ;)
Olaa.. Estava aqui a passear na net, mais precisamente a procurar esse bicho mau chamado 'Huntington' , nao como curiosidade, pois tenho um caso em casa -a minha mae-, mas para me informar ainda mais. E encontrei o teu blog, digo-te ja que fiquei bastante surpreendida, e agradecida, por te dares ao trabalho de escreveres coisas que pra maioria das pessoas nao importam, mas que pra mim soube tao bem ler. Beijinho . Mercês (se quiseres passa no meu www.1brokenstrings.blogspot.com )
Enviar um comentário
<< Home